Ongoing/Previous Research:
- Vitamin D Receptor (VDR) Signaling and Neurodevelopment:
A major focus of my research program has been to define the role of vitamin D receptor (VDR) signaling in neurodevelopment and its susceptibility to environmental perturbation. Building on foundational work in receptor biology, my laboratory has established VDR as a critical molecular interface linking environmental exposures to developmental outcomes in the nervous system. Through a combination of high-throughput screening and in vivo functional models, we were among the first to demonstrate that VDR is a promiscuous target for structurally diverse environmental chemicals, greatly expanding the scope of compounds capable of perturbing neurodevelopmental signaling pathways.
To translate receptor-level interactions into organismal outcomes, my group has leveraged zebrafish as a powerful model of vertebrate neurodevelopment. Using integrative approaches that combine behavioral phenotyping, transcriptomics, and neuroanatomical imaging, we have demonstrated that developmental disruption of VDR signaling leads to persistent and reproducible alterations in neuronal circuitry. These changes manifest as measurable behavioral phenotypes, including deficits in locomotor activity, se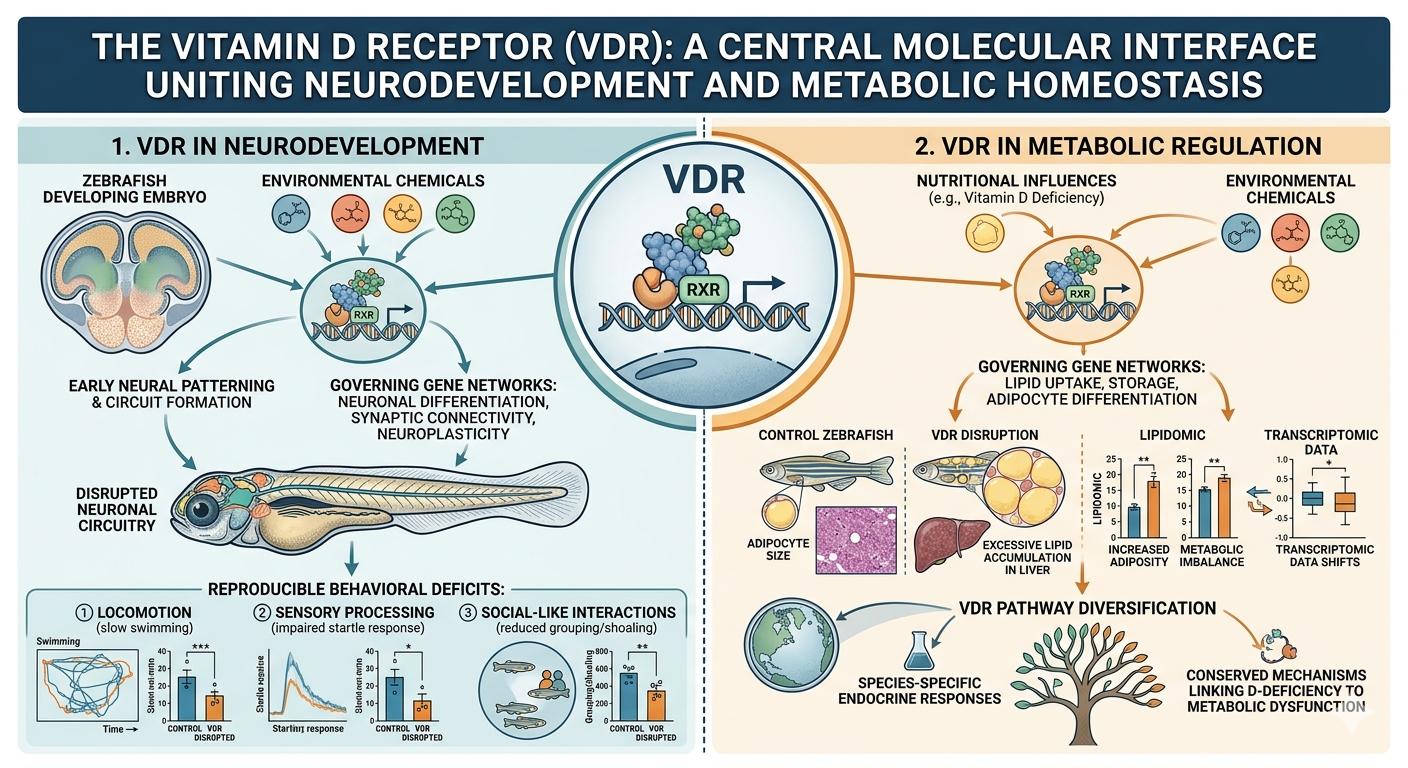 nsory processing, and social-like behaviors. Importantly, these findings provide a direct mechanistic link between chemical:VDR interactions identified in vitro and functional neurodevelopmental outcomes in vivo.
nsory processing, and social-like behaviors. Importantly, these findings provide a direct mechanistic link between chemical:VDR interactions identified in vitro and functional neurodevelopmental outcomes in vivo.
Our work has further revealed that VDR signaling plays a previously underappreciated role in early neural patterning and circuit formation. Perturbation of this pathway during critical developmental windows produces long-lasting effects on gene expression networks governing neuronal differentiation, synaptic connectivity, and neuroplasticity. These discoveries are particularly significant given their relevance to human neurodevelopmental disorders, including autism spectrum disorder and related conditions characterized by early-life environmental influences.
Collectively, this body of work establishes VDR not only as a key regulator of neurodevelopment but also as a sensitive molecular target through which environmental chemicals can exert lasting neurological effects. By bridging molecular, cellular, and behavioral levels of analysis, my research provides a mechanistic framework for understanding how disruptions in vitamin D signaling contribute to neurodevelopmental vulnerability. These insights have important implications for chemical risk assessment, highlighting the need to consider neurodevelopmental endpoints in evaluating VDR-active compounds and underscoring the broader impact of environmental exposures on brain health
- Vitamin D Receptor (VDR) signaling and Metabolic Disruption:
Another central theme of my research has been to elucidate the role of vitamin D signaling in metabolic regulation and its contribution to obesity and cardiometabolic disease. My laboratory has integrated comparative biology, functional genomics, and systems-level metabolic profiling to uncover how perturbations in VDR signaling influence lipid homeostasis across vertebrate species. Early evolutionary studies from my group demonstrated that diversification of the VDR pathway has shaped species-specific endocrine responses, providing a critical framework for understanding variability in metabolic susceptibility and environmental responsiveness.
Building on this foundation, we have employed zebrafish models alongside human clinical data to identify conserved mechanisms linking vitamin D deficiency to metabolic dysfunction. Through the application of lipidomics and transcriptomics, we have characterized distinct metabolic signatures associated with impaired VDR signaling, revealing disruptions in pathways governing lipid uptake, storage, and mobilization. These alterations are accompanied by changes in adipocyte differentiation and energy balance, ultimately predisposing organisms to increased adiposity and metabolic imbalance.
Importantly, our findings challenge the traditional view of vitamin D status as merely a biomarker of metabolic health. Instead, our work supports a model in which VDR signaling acts as a mechanistic driver of metabolic homeostasis. Disruption of this pathway, whether through nutritional deficiency or environmental chemical exposure, leads to coordinated changes in gene networks that regulate lipid metabolism and energy utilization. These effects are remarkably conserved across species, underscoring their biological significance and translational relevance to human health.
In parallel, our discovery that VDR is a target of environmental chemicals has expanded the conceptual framework for obesity research. By demonstrating that environmental ligands can modulate VDR activity and downstream metabolic pathways, we have identified a novel mechanism through which environmental exposures may contribute to obesity risk. This work has important implications for understanding the etiology of metabolic disease in modern environments, where chemical exposures may interact with nutritional status to influence disease susceptibility.
Together, these studies position vitamin D signaling at the center of metabolic regulation, linking environmental exposures, endocrine function, and obesity risk. My research provides a mechanistic basis for integrating vitamin D biology into models of metabolic disease and highlights the importance of considering both nutritional and environmental determinants of metabolic health.
- Aryl Hydrocarbon Receptor (AhR) and stem cell differentiation:
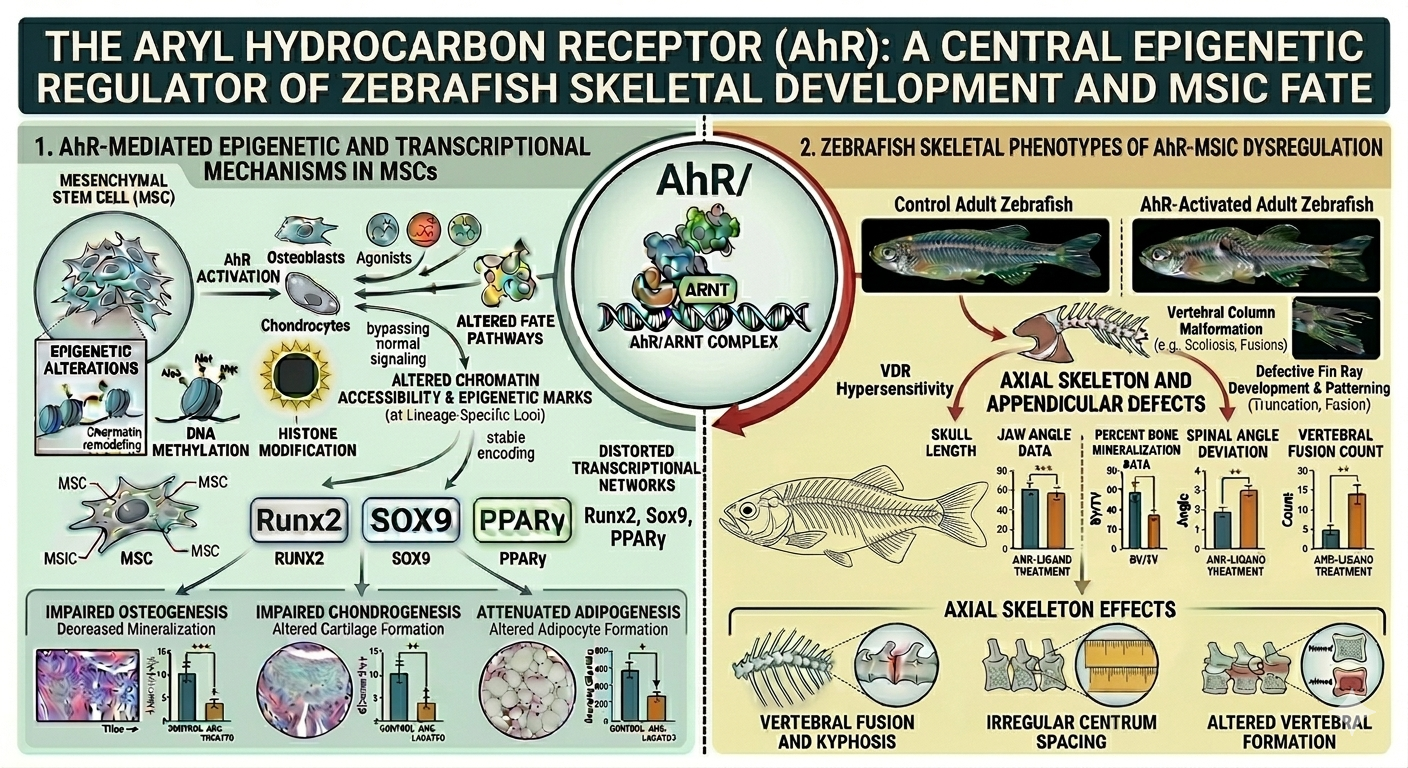
A major focus of my research program has been to define the role of the aryl hydrocarbon receptor (AhR) as a regulator of skeletal biology through its control of mesenchymal stem cell (MSC) fate decisions. Building on the established function of AhR as an environmentally responsive transcription factor, my laboratory has uncovered its previously underappreciated role in directing stem cell differentiation programs that are essential for skeletal development, maintenance, and repair. Our work positions AhR as a central molecular interface through which environmental signals are integrated into the regulatory networks governing MSC lineage commitment.
Using a combination of functional genomics, epigenomic profiling, and mechanistic studies in MSC models, we have demonstrated that AhR activation profoundly disrupts normal differentiation trajectories. Specifically, activation of AhR by endogenous ligands or environmental toxicants alters the balance between osteogenic, chondrogenic, and adipogenic differentiation. These shifts are not merely transient but are likely accompanied by stable changes in chromatin architecture, including altered accessibility at lineage-specific enhancers and promoters. Through approaches such as transcriptomic analysis, we have shown that AhR signaling reprograms the epigenetic landscape of MSCs, effectively biasing lineage commitment away from canonical skeletal pathways.
A key contribution of our work has been the identification of novel transcription factors controlling stem cell fate. These transcriptional alterations disrupt the activation of gene networks required for osteoblast and chondrocyte differentiation while, in some contexts, promoting alternative or dysregulated lineage outcomes. Importantly, these findings provide a mechanistic explanation for how transient environmental exposures can result in long-lasting effects on stem cell function and tissue integrity. By linking receptor activation to durable epigenetic reprogramming, our studies are focused on revealing how environmental information becomes biologically embedded within stem cell populations.
This research has significant implications for skeletal biology and disease. Impaired MSC differentiation directly affects bone formation, cartilage integrity, and adipose deposition within the marrow microenvironment, processes that are critical for bone density, structural integrity, and regenerative capacity. Our findings suggest that persistent AhR activation, particularly in the context of chronic environmental exposures, may contribute to skeletal abnormalities, reduced bone quality, and compromised tissue repair over the lifespan.
Collectively, this body of work establishes AhR as a key regulator of MSC fate and skeletal homeostasis, expanding its role beyond xenobiotic sensing to include fundamental control of stem cell biology. By integrating environmental toxicology with stem cell epigenetics, my research provides a unifying framework for understanding how environmental exposures influence skeletal health and offers new avenues for therapeutic intervention targeting stem cell regulatory pathways.
Recent Comments